Learn more about abnormalities of the auricle’s shape.
Abnormalities of the auricle’s shape
Author: Ian Suchet1, Janelle Santos2
1. Ian Suchet MBBCh FRCPC, Calgary MFM Centre, EFW Radiology, Calgary, Alberta Canada
2. Mayo Clinic, Department of Obstetrics and Gynecology, Rochester, MN, USA
Reviewers: Karen Fung-Kee-Fung, Mauro Schenone
View the Patient Information leaflet
Abnormal auricular shape
Abnormal auricular shape
- Potter’s ear - oligohydramnios
- Lop ear
- Cup ear
- Cleft ear
- Cauliflower ear
- Question mark ear
- Helical deformity - Thick helices, absent helix, absent antihelix, Mozart ear and Stahl ear..
- Lobe / lobular defects.
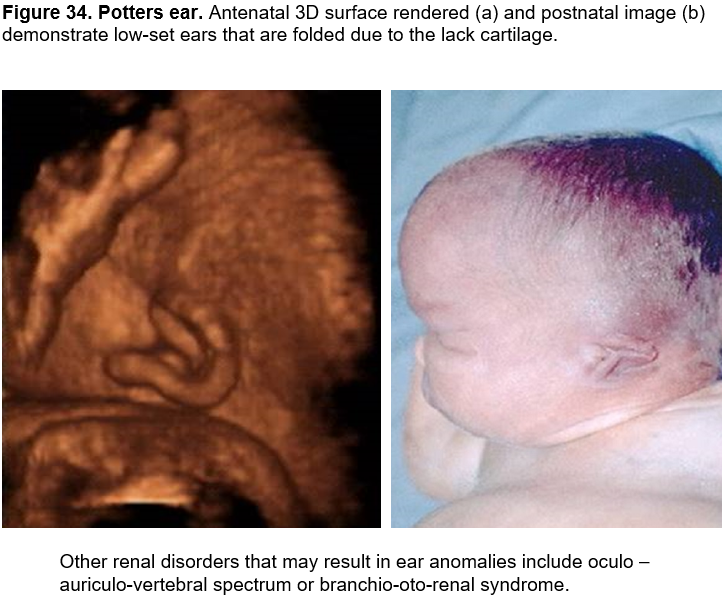
Potter’s ear - oligohydramnios
Potters ear (renal disease): The term "Potter sequence" or "oligohydramnios sequence" is used to describe this condition.
Potters ears may arise secondary to either severe oligohydramnios or anhydramnios (Potters sequence) from any cause.
Originally described by Potter (1) in association with bilateral renal agenesis, Potter sequence is now known to be extremely heterogeneous, both etiologically and pathogenetically (2,3).
Potter sequence is characterized by compression deformities of the face and limbs, pulmonary hypoplasia, and wrinkled skin resulting from any pathologic condition that leads to oligohydramnios including bilateral renal agenesis, cystic renal dysplasia, autosomal recessive polycystic kidneys, bilateral urinary tract obstruction and, persistent amniotic leakage.
Reported ear anomalies include:
- low-set ears
- posteriorly angulated ears,
- a large and floppy auricle due to cartilaginous deficiency. Overfolding of the superior rim of the helix and other alterations in the cartilaginous auricle are frequently caused by fetal constraint (Figure 34).
- a prominent semicircular skin fold that extends from the inner canthus onto the cheek is often seen clinically. When this fold is absent, some functioning kidney tissue may be present (4).
- the ear may be flattened against the head by oligohydramnios, and prolonged pressure against the auricle may result in overgrowth. Enlarged (Macrotia) flattened ears have been associated with renal agenesis as part of Potter sequence. When one ear is exposed to more pressure than the other, the ear anomalies may be asymmetric.
Other craniofacial features include, ocular hypertelorism, blunted nose, micrognathia, and cleft lip and/or palate have been noted in a few instances.
References:
- Potter EL: Bilateral renal agenesis. J Pediatr 1946;29:68-76.
- Passarge E, Sutherland JM: Potter's syndrome. Am J Dis Child 1965;109:80-84.
- Prueger MV et al: Trisomy 7 and Potter syndrome. Clin Genet 1984; 25:543-548.
- Weerda H. Classification of congenital deformities of the auricle. Facial Plast Surg 1988;5:385–388.
Lop ear
Lop ear is an anomaly of the superior one‐third of the auricle and occurs as the superior limb of the antihelix or fossa fails to develop properly. The ear is overfolded in a lop configuration – superior edge of helix and scapha are folded down. The superior crus of the antihelix is either small or absent, the scapha is small, and the auricle is shorter in vertical height. There is often associated exaggeration or overdeveloped of the concha. Without the support of the fully formed cartilage (antihelix) and depending on the extent of the maldevelopment of the antihelical fold, the top of the ear folds over to a varying degree.
Pathology
Davis (1) described hillock 3 hypoplasia as a major factor in the pathogenesis of lop ears. Hillock 3 hypoplasia results in forward displacement of the helix to bridge the gap and the resultant pull causes the superior one third of the pinna to over‐fold. This defect probably represents a continuum of alterations of plical folding of the auricle. It could on some occasions be the result of deforming forces and at other times be a true alteration of the morphogenesis (malformation) of intrinsic muscles and/or cartilage folding.
Frequency
The frequency is thought to be approximately 1:1000-2000 (2).
Frequency based on ethnicity:
-
- Caucasian 2-5%
- African‐descent 10-15%
- There is a significant higher frequency in the Japanese population. Matsuo and colleagues (3) reported that the frequency of lop ear was 38.1% at birth and 6.1% at 1 year of age.
Etiology
A precise etiology or pathogenesis of the lop/cup ear defect is not determined in most individuals who have the defect. Milder degrees of the lop/cup ear defect may truly be a deformation related to intrauterine compression. The fact that many of the cases in Japan of the so-called lop/cup ear defect resolved supports this notion, as does the recent success in tape attachment and other nonsurgical approaches to these defects.
Smith and Takashima (4) and Zerin et al (5) have suggested that the lop/cup ear defects as well as the protruding ear may be due to alterations of plical folding of the cartilaginous ear plate that are, in fact, secondary to changes in the extrinsic and intrinsic ear muscles. These investigators suggest that the ear muscles and perhaps the nerve that innervates the muscles are crucial in determining the form and position of the cartilage of the ear. Their dissections of fetuses with anencephaly, who often have the lop/cup defects, along with experimental evidence in rodents and rabbits suggest that the intrinsic auricular muscles are important for the complicated development of the external ear.
Karmody and Annino (6) described lop ear as an anomaly of the cartilage.
Manigila and Maniglia (7) ascribed it to failure of unfolding of the helix during the 12–16th weeks of intrauterine life
Non syndromic lop/cup ear defects have been seen as an autosomal dominant trait in a number of families; however, the proportion of cases due to an autosomal dominant gene is unknown.
Classification
The line of separation between the lop/cup ear and microtia type I is hard to draw, and some ears described as showing type I microtia fall into this group.
Hunter and Yotsuyanagi (8) sub classified lop ear into a milder form and a moderate form.
- The milder form includes an anomaly limited to helix folding above Darwin’s tubercle.
- In the moderate form of lop ear, the folding of the helix extends below Darwin’s tubercle.
Tanzer (9) has called this defect a “constricted ear” and has divided it into three groups according to the surgical challenges:
Group I defects represent the more traditional lop ear and involve an overfolding of the helix along its superior rim, producing some decrease in height and a flattening of the superior helix / scapha region.
Group Il defects involve both the helix and the adjoining scapha. The lack of cartilage folding involves the crura of the antihelix and the antihelix itself may be flattened. It is sometimes called a cup ear and has components of both lop ear and protruding ear.
Group Ill defects are a severe version of group Il, as the auricle is markedly rolled over the inferior portion of the ear, so the ear is always low-set. This has occasionally been referred to as a “cockleshell ear”.
Associated anomalies
Lop ear can occur as an isolated anomaly.
Associated syndromes in which lop/cup ear may be present (2) include (10):
- Brachio-oto-renal syndrome (branchial sinus, ear and renal anomalies).
- Familial blepharophimosis (blepharophimosis, ptosis, epicanthus inversus).
- Fraser syndrome (cryptophthalmia, syndactyly, Mullerian defects and renal agenesis).
- Kabuki syndrome (Distinctive face, cleft palate, short stature).
- LADD syndrome (lacrimal, auricular, dental anomalies, digital anomalies).
- Lee syndrome (Distinctive face, short stature, premature aging).
- Mengel syndrome (Ear defects, conductive hearing loss, hypogonadism, mental retardation).
- Oto-facio-cervical syndrome (long face, narrow nose, web neck, ear pits, cervical fistulae).
- Townes-Brocks syndrome (imperforate anus, triphalangeal thumb, other digital anomalies, hearing loss).
- Tricho-rhino-phalangeal syndrome (prominent nose, epiphyseal dysplasia).
- Trisomy 21.
- Del 4q (cleft palate, nail defect of second digit, mental retardation).
- Associated with: anencephaly, microcephaly, severe congenital neuromotor deficiency and ossicular malformation.
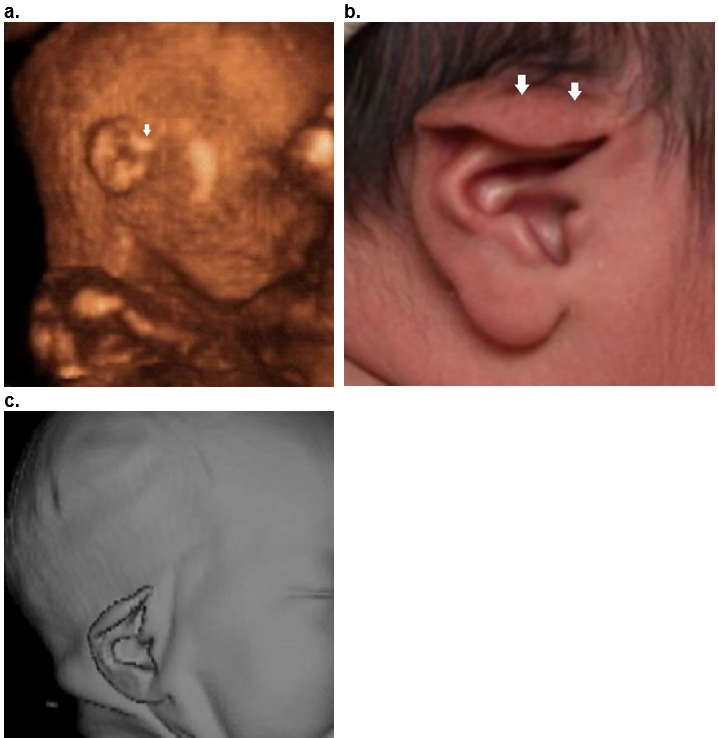
Figure 35. Isolated lop ear.
Case 1.
a. Mild form as only the superior helix is overfolded (closed arrow).
b,c .Postnatal image (b) and postnatal 3D CT scan (c) demonstrates overfolding of the superior helix. The superior crus of the antihelix is obscured by the overfolded helix.
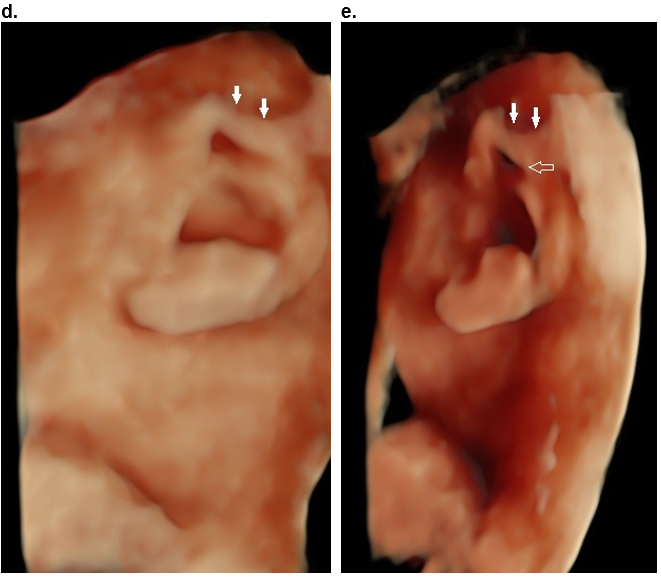
Case 2
d,e. Moderate folding of the superior helix (closed arrows) and the proximal portion of the descending helix (open arrow). The folding of the helix, is more evident when the volume is rotated into an oblique position (e).
References
- Davis J. 1987. Surgical embryology. In: Aesthetic and Reconstructive Otoplasty (ed. J Davis). New York: Springer Verlag, pp. 93–125.
- Stevenson RER, Hall JG. Human malformations and related anomalies 2005. Oxford University Press.
- Matsuo K, Hayashi R, Kiyono M et.al. Non surgical correction of congenital auricular deformities. Clin Plast Surg 1990;17:383.
- Smith DW, Takashima H: Ear muscles and ear form. Birth Defects Orig Artic Ser XVI 1980;(4):299.
- Zerin M, Van Allen MI, Smith DW: Intrinsic auricular form. Pediatrics 1982;69:91.
- Karmody CS, Annino DJ J. Embryology and anomalies of the external ear. Facial Plast Surg 1995;11(4): 251–256.
- Maniglia AJ, Maniglia JV.Congenital lop ear deformity. Otolaryngol Clin North Am 1981;14(1): 83–93.
- Hunter AG, Yotsuyanagi T. The external ear: more attention to detail may aid syndrome diagnosis and contribute answers to embryological questions. Am J Med Genet 2005; 135(3): 237–250.
- Tanzer RC: The constricted (cup and lop) car. Plast Reconstr Surg 1975;55:406-415.Carey JC, Park AH, Muntz HR. 2006. Lop/cup ear anomaly. In: Human Malformations and Related Anomalies (eds Stevenson RE, Hall JG, Sulik KK, Gilbert‐Barness E, Buchanan KK). Oxford: Oxford University Press, pp. 344–346.
Cup ear
Cup-Cupped ear – this is considered an advanced form of a prominent ear with an incompletely formed EAC. Stiff resistant cartilage is present around the scapha and helical rim.
Rogers (1) described it as a deformity with features of both protruding ears and lop ears. In cup ears the protrusion results from overdevelopment of the concave concha. A shorter helix and abnormal antihelix are a few of the common features it shares with lop ears. Due to the forward cupping of the lobule, the vertical height of the ear from the superior pole to the inferior lobe is less than in a normal ear.
Peterson and Schminke (2) published a study of a family with 12 members from five generations with cup‐shaped ears. In addition to cup ears, the proband had Pierre Robin syndrome.
Classification
The cup-ear deformities described by Weerda (3) following Tanzer (4) are classified as follows:
- Type I (slight deformity, corresponding to dysplasia grade I, affects only the helix). This is also referred to as a lop ear. A slight cap-shaped projection of the helix hangs over the scapha; the inferior crus of the antihelix is normally present. The longitudinal axis of the pinna is slightly shortened. Often there is concomitant prominence of the ear.
- Type II deformity (the helix, the anthelix with its crura and the scapha are affected).
- Type IIa (slight to moderate deformity, dysplasia grade I shows a hood-like overhang of the helix accompanied by flattening or absence of the superior crus of the antihelix and a pronounced inferior crus of the antihelix. The shortening of the longitudinal axis of the pinna is greater. The ear is often prominent.
- In type IIb (moderate to strong deformity, dysplasia grade I in the hood-shaped helix overhang and the shortening of the longitudinal axis is more marked. The ear is decreased in width, particularly at the upper part. The crura of the anthelix and the anthelix itself are flattened or absent. The pinna is prominent and straightening usually reveals insufficient skin (Figure 36).
- Type III (severe deformity, dysplasia grade II shows marked underdevelopment of the upper pinna, extreme overhanging of the superior auricular components and considerable deficits in height and width of the ear. There is often dystopia, showing low and anterior positioning, and EAC stenosis is frequently present, occasionally EAC atresia.
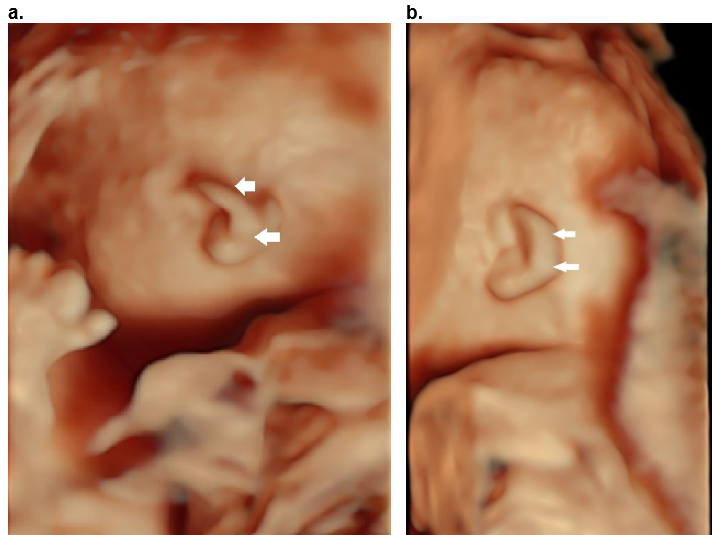
Figure 36. Cup ear deformity type II b.
Hood shaped helix (arrows) with shortening predominantly of the width of the ear. There is overhanging of the superior and descending helix (arrows).
References
- Rogers BO. 1968. Microtic, lop, cup and protruding ears: four directly inheritable deformities? Plast Reconstr Surg 1968;41(3): 208–231.
- Peterson DM, Schimke RN. Hereditary cup‐shaped ears and the Pierre Robin syndrome. J Med Genet 1968;5(1): 52–55.
- Weerda H. Classification of congenital deformities of the auricle. Facial Plast Surg 1988;5:385–388.
- Tanzer RC. Microtia. Clin Plast Surg 1978;5:317–336.
cleft ear
Helical cleft / crease
Congenital cleft earlobe is a rare clinical entity that results from failure of fusion during embryologic development. A helical cleft is seen as a defect in the continuity of the helix, which may occur at any point along its length. This may take the form of a sharp cleft or a less well-demarcated area in the helix. When the defect of the helical fold lies at the junction of the superior and descending portions of the helix, this defect should be coded as Darwin notch of the helix (Figure 37 a,b).
They are more prevalent among boys than girls (2:1).
Usually sporadic, however familial clefts have been reported (2).
Clefts have been described in Treacher - Collins syndrome, however the ears are small and deformed (3).
A helical cleft may be present in CHARGE syndrome (Figure 39 b, d).
An earlobe crease is a line or indentation on the skin surface of the pinna that is otherwise smooth. The crease usually extends diagonally across the lobe from the tragus to the posterior edge of the auricle (figure 37 c,d).
Transverse creases may be present in Beckwith Wiedemann syndrome (Figure 37 c) while a complete cleft is a rare presentation (Figure 51 a,b).
Classification
- Helical clefts have been classified into four types according to morphology by Kitayama et al. (4) – longitudinal clefts, transverse clefts, triple lobes, and defective lobes.
- Yamada et.al.(5) classified them as anterior, posterior and double lobe types.
Ultrasound
A suspected cleft must be evaluated in multiple planes to ensure the cleft is real.
- Unilateral (usually) but may be bilateral.
- Partial or complete clefts. There is a wide range of severity from simple notching to a deep cleft with significant tissue deficiency. Mild indentations that do not involve the entire thickness of the auricle are called creases.
- The is a soft tissue deficiency at the level of the cleft with a curved cleft margin.
- If the cleft is located between the lobule and the lower part of the helix, it may be accompanied by a prominent or deficient upper part of the helix (Figure 37 d), a shallow skin dimple on the posterior surface of the ear, or transposition of the ear lobe/antitragus.
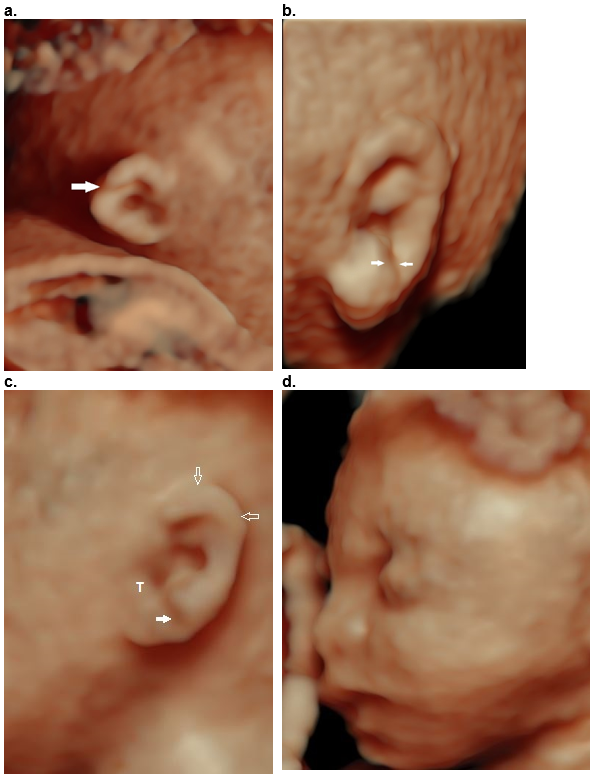
Figure 37. Helical clefts (a,b) and creases (c,d) in four different patients.
- Transverse incomplete helical cleft at the level of the Darwinian tubercle (arrow).
- Oblique complete cleft between the descending portion of the helix and the lobe (arrows) with a normal upper helix.
- Partial crease between the descending portion of the helix and the lobe extending from the tragus (T) to the posterior ear. The superior and descending helix are normal (open arrows). This was a case of Beckwith – Wiedemann syndrome.
- Partial crease between the descending portion of the helix and the lobe with a deficient superior and descending helix (open arrows).
Both clefts (a,b) were isolated and non syndromic. The crease in (c) was due to Beckwith – Wiedemann syndrome, and no syndrome was found for the crease in (d).
References
-
- Hunter A, Frias JL, Gillessen-Kaesbach G, Hughes H, Jones KL, Wilson L.. Elements of morphology: Standard terminology for the ear. Am J Med Genet Part 2009;149A:40–60.
- Uppal S, Garg R. Recurrent cleft of the ear lobe in siblings of the same family. Int J Plastic Surg 2008;6:
- Davis J. Otoplasty aesthetic and reconstructive techniques. Springer, New York 1999.
- Kitayama Y, Yamamoto M, Tsukada S. Classification of congenital cleft earlobe. Jpn J Plast Reconstr Surg 1980;23:663-70.
- Yamada A, Fukuda O, Soeda S, et al. The evaluation of cleft earlobe. Jpn J Plast Reconstr Surg. 1967;19:171–175.
Cauliflower ear
Diastrophic dysplasia is a rare autosomal recessive inherited osteochondrodysplasia – intrinsic abnormalities of cartilage and bone. The characteristic clinical features result from impairment of physeal, epiphyseal and articular cartilage due to undersulfation of proteoglycan in the cartilaginous matrix.
The underlying genetic basis of diastrophic dysplasia is the result of a mutation in a novel sulfate transporter gene, known as the diastrophic dysplasia sulfate transporter (DTDST) and SLC26A2 (1). Impaired function of this gene product leads to undersulfation of proteoglycans in cartilage matrix, which leads to abnormal cartilage formation and results in the disease phenotype (1).
It is characterized by micromelia, cleft palate, kyphoscoliosis, limited joint mobility, talipes, varus deformity of the great toes and distinctive abduction of the first metacarpals (‘hitchhiker’ thumbs). The ear abnormality consists of cystic degeneration of the pinna (1,2).
Eighty five percent of infants with diastrophic dysplasia develop cystic ear swelling usually in the neonatal period but this abnormality has been reported antenatally. This type of ear involvement is a pathognomonic feature of the disorder. The auricles typically develop acute, recurrent, bilateral fluctuant swelling which normally resolves over a 3–4 week period and is replaced with thickening and deformity of the auricular cartilage with progression to “cauliflower” ear and ultimately calcification and ossification of the auricle by adulthood (Figure 38).
There may be associated narrowing of the EAC.
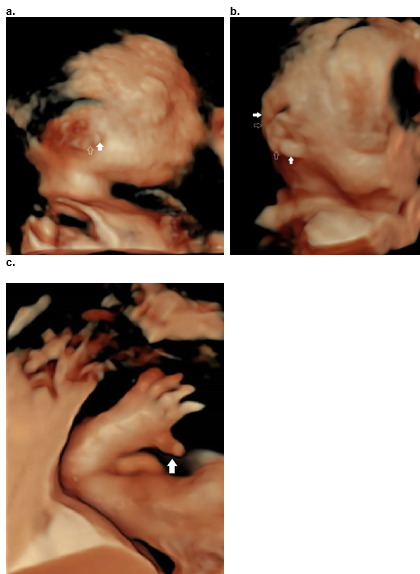
Figure 38. Diatrophic dysplasia.
a.b. Cauliflower ear with focal areas of swelling (closed arrow) and interspersed areas of narrowing / stricturing representing areas of fibrosis (open arrow).
c. Hitchhikers thumb.
References
- Unger S, Superti-Furga A. Diastrophic Dysplasia. 2004 Nov 15 [Updated 2021 Dec 23]. In: Adam MP, Everman DB, Mirzaa GM, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993-2022.
- Hastbacka J, de la Chapelle A, Mahtani MM et.al. The diastrophic dysplasia gene encodes a novel sulphate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell 1994;78(6):1073-1087.
Question mark ear
The Cosman - Question mark ear (Figure 39) is an auricular abnormality characterized by a cleft between the lobe and the lower part of the helix (between the 5th and 6th hillocks), sometimes accompanied by a prominent or deficient upper part of the helix, a shallow skin dimple on the posterior surface of the ear, or transposition of the ear lobe/antitragus. The upper portion of the ear appears protruding and the scapha is absent from the affected region (1,2).
Classification
Pan et al. (3) published a 32‐case series which included 30 sporadic cases and two of familial origin. They divided the ear defect into moderate and severe types depending on the anatomical characteristics. In the moderate type the lobule and inferior part of the helix presented with a cleft, and in the severe type the lobule and inferior part of the helix were absent.
Ultrasound
Male> female
Unilateral or bilateral.
The cleft varies from a small notch (figure 39 d) to complete separation of the helix from the lobe.
The helix is normal (Figure 39 a) or deficient (Figure 39 b).
Associated anomalies
The ear deformity is seen clinically in three different presentations:
- sporadic, familial (usually autosomal dominant).
- isolated ear deformity.
- a constant – pathognomonic feature of aurico-condylar syndrome which is a rare craniofacial disorder with mandibular hypoplasia, microstomia, TM joint abnormalities and question-mark ears as major features.
Sporadic cases may be bilateral or unilateral (4), while inherited cases are almost always bilateral. Most patients with familial isolated ear deformities have mutations of the ENDOTHELIN-1 (EDN-1) gene (5). Most patients with aurico-condylar syndrome have mutations of the PHOSPHOLIPASE C-Beta 4 (PLCB4) gene (6).
Other syndromes include Townes‐Brocks syndrome, Treacher‐Collins syndrome, and oculoauriculo‐vertebral spectrum.
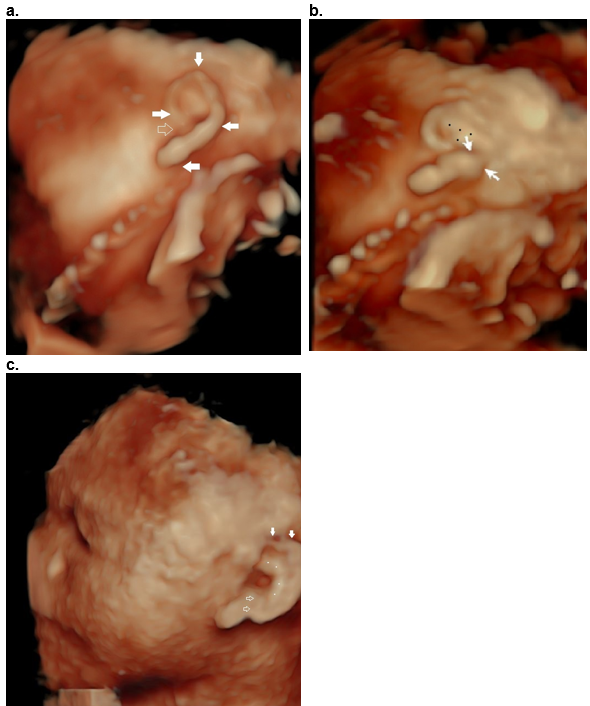
Figure 39. Question mark ear as an isolated deformity.
Case 1
a,b. Question mark shaped outer helix and lobe (closed arrows, a), with an open notch (open arrow, a). The incomplete cleft (closed arrows, (b) between the decending helix and lobe is only visualized when the image is rotated into an oblique position (b). There is a Y shaped antihelix (black dots, b).
Case 2
c. Question mark ear in CHARGE syndrome. Hypoplastic superior helix (closed arrows) and cleft between the lobe and descending helix (open arrow). The antihelix is abnormal (white dots).
References
- Cosman B. The question mark ear: an unappreciated major anomaly pf the auricle. Plast Reconstr Surg. 1984;73:572–576.
- Cosman B, Bellin H, Crikelair GF. The question mark ear. Plast Reconstr Surg. 1970;46:454–457.
- Pan B, Jiang H, Zhao Y, Lin L, Guo D, Zhuang H. Clinical analysis, repair and aetiology of question mark ear. J Plast Reconstr Aesthet Surg 2010;63(1): 28–35.
- Al-Qattan MM. Cosman (question mark) ear: congenital; auricular cleft between the fifth and sixth hillocks. Plast Reconstr Surg 1998;102:439–441.
- Gordon CT, Petit F, Kroisel PM, et al. Mutations in endothelin cause recessive auricocondylar syndrome and dominant isolated question-mark ears. Am J Hum Genet. 2013;93:1118–1125.
- Kido Y, Gordon CT, Sakazume S, et al. Further characterization of a typical features in auricocondylar syndrome caused by recessive PLCB4 mutations. Am J Med Genet. 2013;161A:2339–2346.
Helical deformity - Thick helices, absent helix, absent antihelix, Mozart ear and Stahl ear
Absent upper helix (Figure 40 a) – The superior and descending helix is absent (arrows). There is thickening of the stem of the antihelix (**) with absent superior and descending portions. A wide concha (8) is present.
Thickened upper and descending helix (Figure 40 b,c). This finding is thought to be due to a variation in plical folding or an alteration in the intrinsic auricular muscles. This may involve the crus of the helix which flares posteriorly (the crus of the helix usually extends around inferiorly and posteriorly to the concha above the EAM). This may produce an extremely prominent fold that has been described as a “railroad track ear) (1).
Syndromes associated with a prominent helix include fetal alcohol syndrome and Sathre-Chotzen syndrome (1,2).
Absent antihelix (Figure 40 d) – There is no ridge between the concha and triangular fossa and helix. This is commonly found in a protruding or cup ear where the superior and inferior part of the antihelix is absent.
Mozart / Wildermuth ear / anteverted concha (eponym to reports of this deformity thought to be present, but not proven, in Wolfgang Amadeus Mozart’s and likely present in his son Franz).
The Mozart ear is a rare auricular deformity. The auricle is characterized by the bulging appearance of the anterosuperior portion of the auricle due to fusion of the crura of the antihelix, an inversion in the normal form of the cavum conchae resulting in a convex appearance and resultant slit-like narrowing of the orifice of the external auditory meatus (3-6).
It has been speculated that this deformity arises from either external forces or abnormal insertion of the auricular muscles.
It is thought that this ear abnormality has an autosomal dominant trait.
This variation in auricular shape has no clinical significance unless it is associated with a syndrome.
Ultrasound (Figure 40 e,f)
Unilateral or bilateral.
Auricle is broad appearing.
Antihelix: A prominent antihelix tends to jut out and obliterate the curve of the helix (two crura of the antihelix are fused and are united with the crus of the helix).
Concha: The concha is enlarged, and the antitragus may be absent or underdeveloped. In a normally developed auricle, conchal cartilage has a concave shape, and this concavity forms the cavum concha cavity. When the conchal cartilage has anterior convexity rather than concavity, this is called “inverted concha”.
EAC: This anterior convexity of the concha results in slit like narrowing of the EAC.
Lobe: The ear lobe may be underdeveloped or very small. In some cases this is so great that the upper half of the ear is flat and there is virtually no helical curve (3-5).
Associated syndromes
Although Davies (5) do not cite the occurrence of this finding as part of a generalized syndrome, prominence of the antihelix is a consistent finding in the deletion 18q syndrome (5,6). A review of photographs of children with 18q- suggests that their finding is also part of the continuum of the Mozart ear.
The so-called faun ear described in the trisomy 18 syndrome also has components of the Mozart ear as well as in CHARGE syndrome (9).
Stahl ear
A Stahl ear is an abnormal extra fold or crus of antihelix that extends from the superior portion of the antihelix to the upper posterior aspect of the helix. This extra fold produces a “crumpled ear” appearance (1).
It is usually isolated (reported in 8% of Japanese newborns) (2) but has been diagnosed as a part of congenital contractual arachnodactyly syndrome (1).
References
- Jones KL. Smith’s recognizable pattern of human malformations. Ed 5. WB Saunders Company, Philadelphis. 1997.
- Matsuo K, Hayashi R, Kiyono M et.al. Non surgical correction of congenital auricular deformities. Clin Plast Surg 1990;17:383.
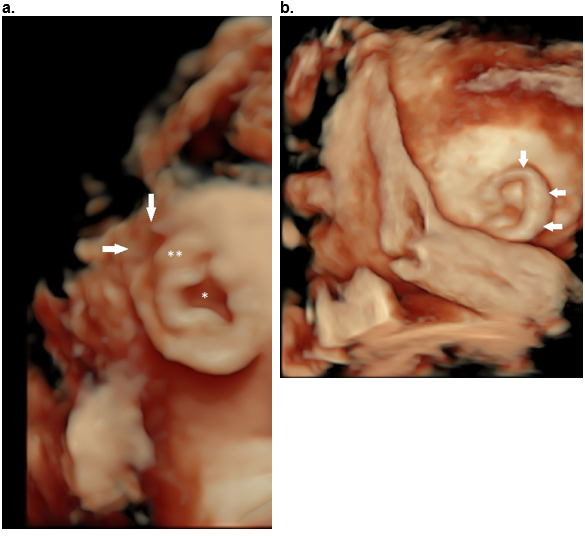
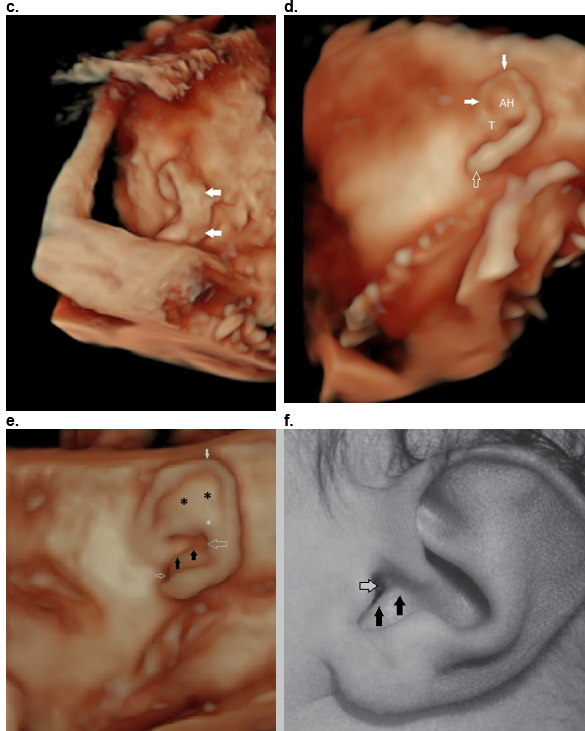
Figure 40: Helical abnormalities.
Absent superior helix (arrows), thickened antihelix (**) and wide concha (*).
b,c. Thick superior and descending helix (arrows).
d. Absent antihelix (AH) in a Question mark ear.
e,f. Mozart ear. Normal stem of the antihelix (white *) with fusion of the two crura of the antihelix (black **). This results in bulging of the superior auricle (closed white arrows). There is anterior convexity of the conchal cartilage (black arrows e,f) that narrows the EAM (small open white arrow e, f). The large white arrow (e) represents the posterior concha.
Reference, postnatal image f: Fletcher MA, ed. Physical Diagnosis in Neonatology. Philadelphia, PA: Lippincott–Raven Publishers; 1998:288).
References
- Aase JM. Microtia-clinical observations. Birth defects Orig Artic Ser XVI 1989;(4):289.
- Jones KL. Smith’s recognizable patterns of human malformations. Ed 5. 1997:WB Saunders Company, Philadelphia.
- Telich-Tarriba JE, Victor-Baldin A, Apellaniz-Campo A. Mozart ear deformity: a rare diagnosis in ear reconstruction clinic. J Craniofac Surgery 2017;28:e482-e484.
- Yamashita K, Yotsuyanagi T, Saito T et.al. Mozart ear: diagnosis, treatment, and literature review. Ann Plast Surg 2011;67:547-550.
- Davies PI: Mozart’s left ear, nephropathy and death. Med Aust 147:581, 1987.
- Paton A, Pahor Al, Graham GR, et al.: Looking for Mozart ears. Br Med J 1986;293:1622-1624.
- Jones KI: Smith's Recognizable Patterns of Human Malformation, ed 5. WB Saunders Company, Philadelphia, 1997.
- Garcia-Cruz D, Sanchez-Corona J, Runes R, et al.: A syndrome with mixed deafness, Mozart ear, middle and inner ear dysplasias. Laryngol Otol 1980;94:773. Stevenson RE, Hall JG. Human malformations
Lobe / lobular defects
Lobe / Lobular defects are alterations of the form or contour of the ear lobe. They are usually unilateral and rarely syndromic.
Congenital anomalies of the lobe are less commonly described and are often characterized in the context of congenital earlobe cleft.
Lobe defects include:
- Absent ear lobe - Complete absence of the lobule is rare, particularly when other auricular components are fully or partially developed.
- Bifid ear lobe.
- Notched ear lobe.
- Uplifted lobes (have been reported in Turner syndrome, probably due to lifting of the lobe by the cystic hygroma rather than the syndrome itself).
- Antitragus base tag.
- Thickened ear lobes - they have been described as an external marker associated with conductive hearing loss and discontinuity between the incus and stapes (1). This finding has been described in two families and is referred to as Escher-Hirt syndrome (2).
References
- Goldberg MJ, Pashayan HM. Hallux syndactyly-ulnar polydactyly abnormal ear lobes: a new syndrome. Birth Defects Orig Artic Ser 1976;12(5):255-266.
- Escher F, Hirt H. Dominant hereditary conductive deafness through lack of incus stapes junction. Acta Otolaryngol 1966;65:25.
This article should be cited as Suchet I, Santos J: Abnormalities of the auricle’s shape . Visual Encyclopedia of Ultrasound in Obstetrics and Gynecology, www.isuog.org, June 2023.
Leave feedback or submit an image
We rely on your feedback to update and improve VISUOG. Please use the form below to submit any comments or feedback you have on this chapter.
If you have any images that you think would make a good addition to this chapter, please also submit them below - you will be fully credited for all images used.
Feedback form
Please note that the maximum upload size is 5MB, and larger images and video clips can be sent to [email protected].